Introduction to JR-Assembler
Method
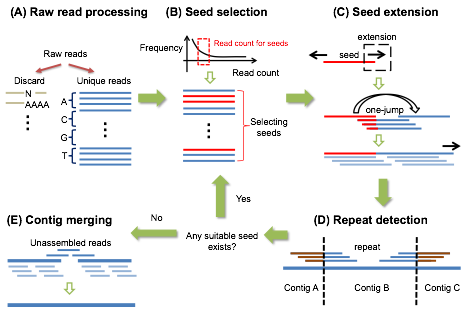
JR-Assembler
runs in five steps: raw read processing, seed selection, seed extension,
repeat detection, and contig merging. First, all reads
containing any base ‘N’ or any low
complexity region are filtered out. Second, it selects “good” reads as
seeds using the read count, i.e., the number
of identical reads in the data. Third,
JR-Assembler uses a “jumping” extension, including many whole
reads at a time. Moreover, to
deal with sequencing errors at read
tails, JR-Assembler uses back trimming to remove low quality nucleotides at the 3'-end
of a read to facilitate extension. Fourth, when an extension is terminated,
JR-Assembler checks whether a mis-extension was made because of the existence of a repeat. If a mis-extension occurs,
it identifies
the boundaries of the repeat and breaks the sequence at the boundaries. The
three
steps of seed selection, seed
extension, and mis-extension
detection are repeated until no unused seed remains. Finally, JR-Assembler takes care of
low coverage regions by applying a less stringent extension procedure to merge the assembled sequences. JR-Assembler also incorporates a scaffolding program, SSPACE
(1), for users to
construct scaffolds.
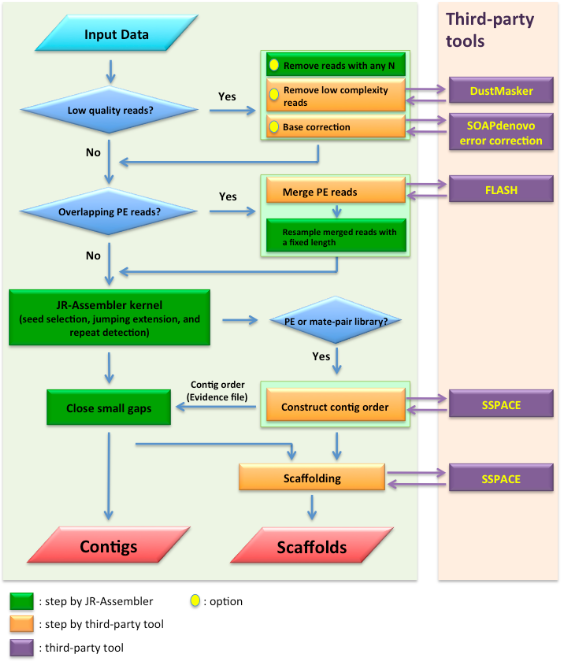
Work flow
of JR-Assembler
For more details of
JR-Assembler, please refer to
Te-Chin Chu, Chen-Hua Lu, Tsunglin Liu, Greg C. Lee, Wen-Hsiung Li, and Arthur Chun-Chieh Shih, “Assembler for de novo assembly of large genomes,” Proceedoings of the National Academy of Science, September 3, 2013 vol. 110 no. 36 E3417-E3424.
(abstract)
(pdf)
|
|
References
1.
Morgulis A, Gertz EM, Schaffer AA,
& Agarwala R (2006). A fast and symmetric
DUST implementation to mask low complexity DNA sequences. J Comput Biol 13(5):1028-1040.
2.
http://soap.genomics.org.cn/soapdenovo.html
3.
Magoc T, Salzberg SL (2011). FLASH: fast length
adjustments of short reads to improve genome assemblies. Bioinformatics 27:2957-2963.
4.
Boetzer M, Henkel CV, Jansen HJ, Butler D,
& Pirovano W (2011). Scaffolding pre-assembled
contigs using SSPACE. Bioinformatics 27(4):578-579.
|
